Our research
Drug discovery is fundamentally a search across vast chemical and biological spaces. Our group develops computational tools at the interface of chemistry, biophysics, and machine learning to navigate these spaces. In turn, these tools are applied to discover new chemical starting points for drug design against viral infections, cancer, and other diseases. In our research, we are fortunate to collaborate with several national and international groups with leading expertise in medicinal chemistry, biology, and pharmacology. Feel free to contact us if you want to know more or explore collaborations.
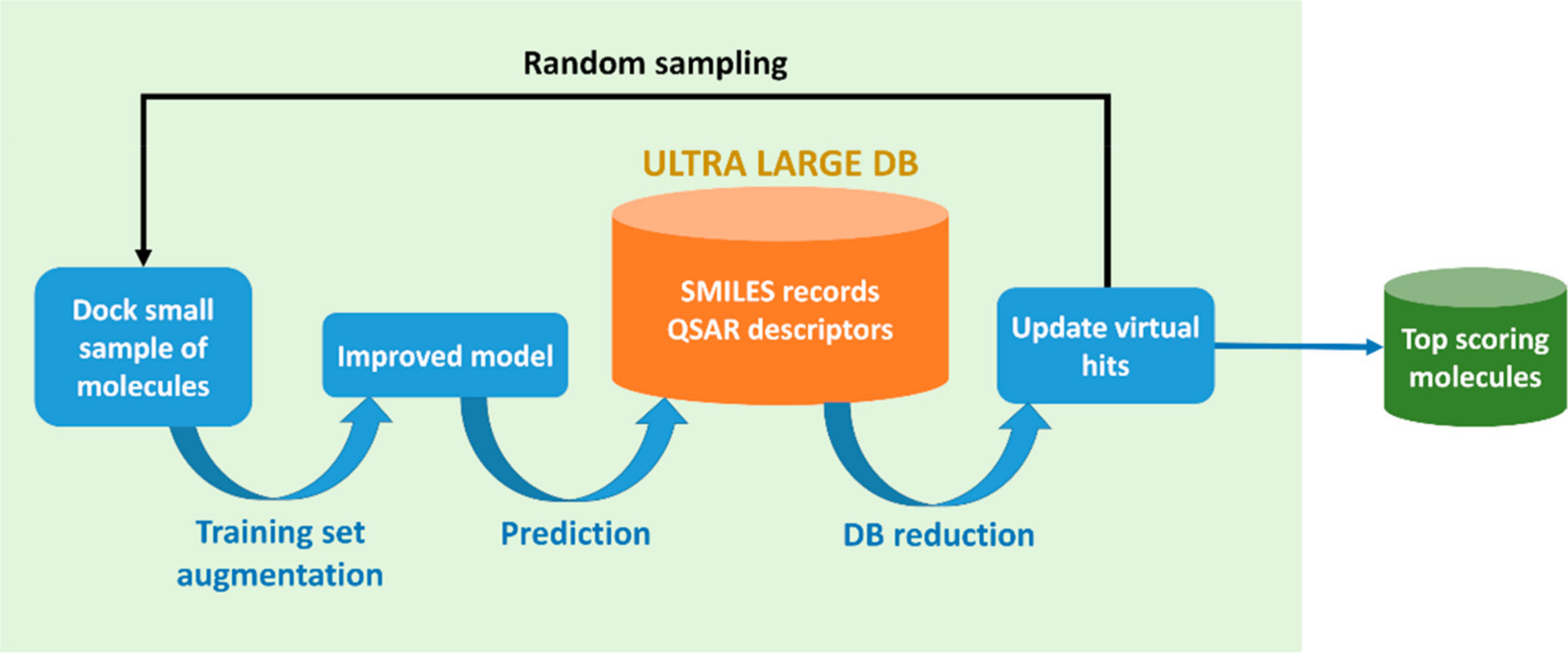
While traditional virtual screening has focused on libraries of up to a few million compounds, the number of possible drug-like molecules exceeds 10⁶⁰, and modern make-on-demand libraries now contain billions of accessible candidates. Exploring this space efficiently remains a major challenge. To address this, we develop machine learning approaches to accelerate structure-based virtual screening by building fast and reliable surrogates of physics-based methods. This enables scalable exploration across ultra-large libraries to investigate their therapeutic potential, and broadens access to high-throughput computational chemical discovery. This review from our lab is a great starting point to learn more about the topic.
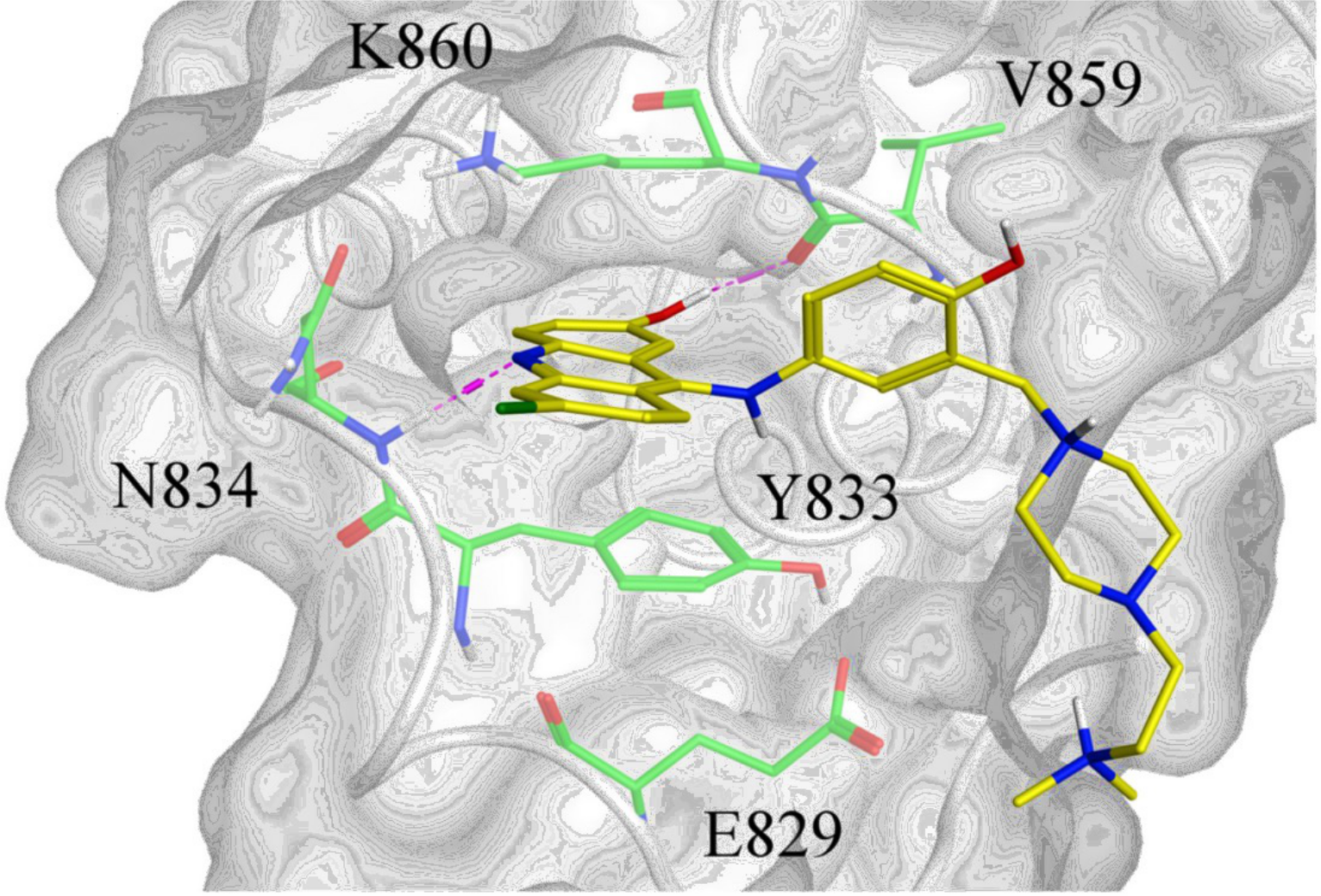
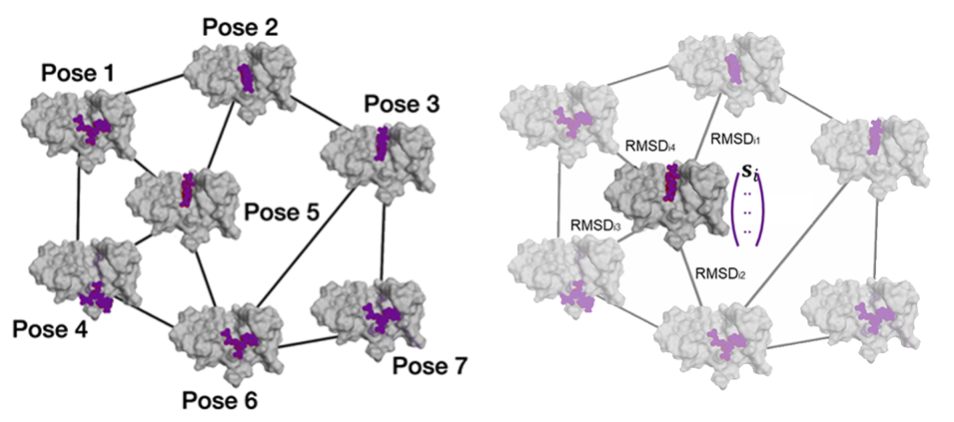
Besides acceleration, we also strive to improve the robustness of virtual screening methods, by addressing artifacts and failures on novel or flexible targets through the integration of data-driven models with physics-based tools, such as in DockBox2 . We are especially interested in improving chemical discovery against challenging, emerging targets such as allosteric sites, transient pockets, protein-protein and protein-nucleic acid interactions, which remain difficult for conventional approaches.
We are also interested in understanding and overcoming drug resistance in cancer and bacterial infections. To this end, we apply computational methods to design small molecules that either disrupt key resistance mechanisms, including inhibitors of DNA damage response pathways that limit the efficacy of chemotherapy in cancer, or probe novel mechanisms that are less susceptible to resistance. Beyond small molecules, we are also interested in improving the design of complex biologics such as antibody–drug conjugates (ADCs), using machine learning to guide discovery and optimization across biological space.